General
Overlap ligation & Gibson ligation
Restriction endonuclease reactions
4HB biosensor construction
We constructed a 4HB biosensor based on plasmid pYB1a-PobR-eGFP-Cmr. This plasmid mainly contains two parts:
1. The pobR coding sequence (CDS) which can express PobR protein;
2. An engineered operon consisting of the enhanced green fluorescent protein (eGFP) and the chloramphenicol resistance (Cmr) genes.
To further understand each part, please click here to view our design part.
To constructed pYB1a-PobR-eGFP-Cmr, we first tried to generate plasmid pYB1a-PobR-eGFP by Gibson assembly. PobR CDS was synthesized by Ruibiotech (Harbin, China) and amplified by PCR using the primers PobR-Gibson-F and PobR-Gibson-R. At the same time, plasmid pYB1a-eGFP was linearized using primers pYB1a-Gibson-F and pYB1a-Gibson-R. Then the two fragments were ligated using Gibson assembly and transformed into E. coli DH5α competent cells and the plasmid pYB1a-PobR-eGFP was constructed. Click here to view our ligation protocol.
Secondly, we tried to insert Cmr gene into pYB1a-PobR-eGFP by using enzyme-cut and Link up. The Cmr CDS contained with the restriction endonuclease cleavage sites of XhoI and BglII was amplified by Cmr-F and Cmr-R primers using the Cmr CDS as a template. Then the fragment and the plasmid were digested with XhoI and BglII. Finally, we used T4 ligase (Thermo Fisher Scientific) to ligate the two fragments and transformed the ligation product into E. coli DH5α competent cells.
The result of DNA sequencing showed that our pYB1a-PobR-eGFP-Cmr plasmid was constructed.
Construction of PobR mutant library
Aiming to get the HMA biosensor with the best performance, a large PobR mutants library which is able to include as many mutation situations as possible is crucial. Therefore, we designed our mutants library construction method based on error-prone PCR and MEGWHOP ( megaprimer PCR of whole plasmid) cloning [1]. This method has high efficiency and high positive fate compared with traditional methods. Please click here to view our detailed protocol.
A large library for stochastic mutagenesis of PobR was established by using error-prone PCR and MEGAWHOP (megaprimer PCR of whole plasmid). Mutations on PobR were performed by random mutagenesis kit (Solarbio Life Science). Random mutation codons were introduced into the amplified targeted pobR gene, due to Taq DNA polymerase lack 3’→5’ proofreading function in the reaction buffer system. It is conspicuous to substantiate that the positions mutated for the library were diversiform, and the mutated rate is 3‰on average by DNA sequencing. The amplified targeted pobR genes were inserted into the template by MEGAWHOP without requiring restriction digestion in these constructs [1]. The enzyme of DpnI (New England Biolabs) was applied into the whole-plasmid PCR products to digest methylated and hemi methylated DNA templets [2].T4 DNA ligase (Thermo Fisher Scientific) was be employed to concatenate incision plasmid for improving the efficiency of transformation. Upon ligation with T4 DNA ligase, PobRmut constructs was obtained. The ligated products were transformed in DH5α competent cell (Tsingke Biotechnology).
Based on the methods introduced above, about 552,750 colonies containing PobR mutagenesis were established, in this PobRmut constructed library.
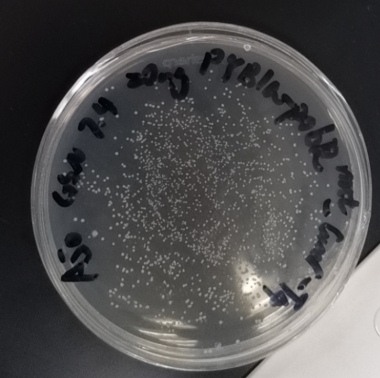
Figure1.1100 colonies grew on the plate after the plasmid with mutated points (20 ng) was transformed in DH5α competent cell (100 μl). 55 colonies were recuperated from per nanogram of DNA.
Fluorescence assay and screening of the PobR mutant library
Another part which is essential to get the best HMA biosensor is the screening part. To quickly and precisely selected clones from our huge mutant library, we used two different methods: chloramphenicol growth screening and fluorescence assay screening to meet our target.
First, we transformed the PobR mutant library into DH5α competent cells and cultured overnight in LB agar plates with 20 μg/mL chloramphenicol as growth pressure and 0.06 g/L HMA as inducer (50 μg/mL ampicillin was also added in LB to remain the plasmid).
In this progress, 639 clones grew on the plates were picked to our second step screening.
The second step of our screening is fluorescence assay. We used Tecan Spark® multimode microplate reader to measure OD600 and fluorescence intensity of enhanced green fluorescence protein (eGFP). By adding 1 g/L HMA as inducer, the clones with higher induction of HMA would show a higher RFU/OD600 value compared with blank control (no HMA was added) and other clones.
We used deep-well 96-well microplates containing 200 μl of LB medium with ampicillin to culture the clones picked from the first step. After shaking at 200 rpm and 37°C for 12h, 2 μl of the bacteria from each well was inoculated into 200 μl of LB medium with ampicillin and 1 g/L of HMA in deep-well 96-well microplates, and cultured on a shaking incubator for another 12 h. Finally, we picked 100 μl of the cultured bacteria from each well into the 96-well microplate to detect the OD600 and green fluorescence (with 430 nm as the excitation wavelength and 510 nm as the emission wavelength). Please click here to see our microplate reader operating procedure.
Ligand specificity test of selected PobR mutants
We used fluorescence assay to detect the ligand specificity of our PobR mutants. Six HMA analogues were used to induce our PobR mutants. The six analogues are MA (Mandelic Acid), HPP (4-Hydroxyphenylpyruvate), PPA (Phenylpyruvate), Trp (Tryptophan), Phe (Phenylalanine), 4HB (4-hydroxybenzoic acid). To study the induction of these analogues to the mutants, we first configurate their mother liquors with the concentration of 50 g/L and pH of 7. Subsequent steps were just like our fluorescence assay screening of the PobR mutant library. We cultured the mutants in 200 μl of LB medium with ampicillin for 12 h at 37 °C. Then, 2 μl of each clone was added to 200 μl of LB medium with ampicillin and 1 g/L of HMA, or each of its analogs (added 4 μl 50 g/L mother liquors), in the wells of a deep-well 96-well microplate, and cultured for 12 h at 37 °C in a shaking incubator. We picked 100 μl of the cultured bacteria from each well into the 96-well microplate to detect the OD600 and green fluorescence (with 430 nm as the excitation wavelength and 510 nm as the emission wavelength). Finally, we calculated the RFU/OD600 of each analogue to each mutant to estimate the mutants’ ligand specificity. Please click here to see our microplate reader operating procedure.
Site-directed mutagenesis of the PobR CDS
To get the PobR mutants with single amino acid mutation, we designed pairs primers with mutation site present in the forward primers to having inverse PCR using pYB1a-PobR-eGFP-Cmr as template [3]. After DNA purification, the original templates were eliminated by DpnI digestion. Finally, the plasmids with nicks on them were transformed into E. coli DH5α competent cells to amplify the plasmids and repair the nicks.
Modeling and docking
To better understand the effects of amino acid substitutions on the response of the PobR protein, we tried to use software to simulate the structure of the protein and construct molecular docking between PobR protein and its inducer.
We first simulated the structure of the PobR monomer by using AlphaFold2 [4] and selected the predicted structure with the highest score (pLDDT = 91.4) from five predictive models which were generated by AlphaFold2 based on the confidence data to have our subsequent molecular docking.
Secondly, we obtained the structure files of the ligand 4HB from the organic small molecule database Pubchem (https://pubchem.ncbi.nlm.nih.gov/).
Based on the structures of PobR monomer and 4HB, molecular docking was simulated by docking engine Autodock Vina [5]. Vina search space coordinates were set as center_x = -0.83, center_y = -1.292, center_z = 7.229. Dimensions of the search space were set as size_x = 33.75, size_y = 37.5, size_z = 37.5, and exhaustiveness was set at 10. The top 9 conformational conditions in a score based on the lowest binding energy (-6.0 kcal/mol) were listed as the docking results. To examine the accuracy of our docking, we used Ligplus to check the interaction between the small molecule and predicted protein receptors [6]. Finally, the three-dimensional schematics of the protein and its ligand were portrayed using PyMol Version 2.2.0.
Adaptive laboratory evolution procedure
To evolve strains with higher HMA production three modules includes HMA biosensor module (plasmid pYB1a-PobR-eGFP-Cmr), HMA biosynthesis module (plasmid pRB1s-hmaS-aroG-pheA), mutagenesis module (plasmid pLT1k-MP6) were transformed into E. coli BL21 competent cells and named as HMA-0. Then, we inoculated 200 μL of M9 medium with 2% volume of E. coli, 2 g/L glucose and different concentrations of chloramphenicol (5, 10, 15, 20, 30, 40, 50 µg/mL) and shaking at 37 ℃ for 12 h.
Then, we stored the bacteria growth under the pressure of 50 µg/mL chloramphenicol and named as HMA-C50-1. This process was repeated using HMA-C50-1 under the pressure of 60, 80, 100, 120, 140, 160 µg/mL chloramphenicol. The HMA-2 was selected under the pressure of 60 µg/mL chloramphenicol.
Then, we gradually raised the chloramphenicol concentration to enrich HMA-producing cells but to eliminate other cells. When strains grow to stationary phase, 2 μL culture was stored and diluted into a fresh medium with same pressure or higher pressure of chloramphenicol compared with the round before, and the enrichment procedure was repeated for eight rounds. During the enrichment procedure, chloramphenicol was added at a concentration of 50, 60, 60, 65, 65, 70, 75 and 80 µg/mL.
HMA determination
We used HPLC (high performance liquid chromatography) as our HMA determination method. We first activate the cells by inoculate 5 mL of LB medium with 1% volume of E. coli culture and shaking at 200 rpm and 37℃ for 12h. Then we inducted the expression of the protein on HMA biosynthesis module using ZYM-5052 medium and we also used fermentation medium M9 to help the cells produce HMA.
Finally, the HMA was measured an HPLC (Agilent 1260) equipped with a UV detector and a Welch Xtimate C8 column (4.6 × 250 mm, 3.5 µm). The samples were analyzed under the following gradient of eluent A (8 mM KH2PO4, pH 2.4) and eluent B (100% acetonitril): 5 min 10% B, 20 min linear gradient to 30% B, 3 min linear gradient to 50% B, 5 min 50% B, 2 min linear gradient to 10% B, 5 min 10% B. HMA was detected at 215 nm. Please click here to see our detailed HMA determination protocol.
Please click here to see our strains, plasmids and primers used in our experiments.
References
- Miyazaki K: MEGAWHOP cloning: a method of creating random mutagenesis libraries via megaprimer PCR of whole plasmids. Methods Enzymol 2011, 498:399-406.
- McClelland M, Nelson M, Raschke E: Effect of site-specific modification on restriction endonucleases and DNA modification methyltransferases. Nucleic Acids Research 1994, 22:3640-3659.
- Liu H, Naismith JH: An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol 2008, 8:91.
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, et al: Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596:583-589.
- Trott O, Olson AJ: AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 2010, 31:455-461.
- Laskowski RA, Swindells MB: LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model 2011, 51:2778-2786.