Lab notebook
This part records the progress of our team and some communication activities we participated in.
Week 1 ~ Week 2
We build our team, and did research for iDEC project direction. Last, we aimed at peptide drug discovery.
Week 3 : 2021/03/23
We came up with an original version of program framework to begin with.
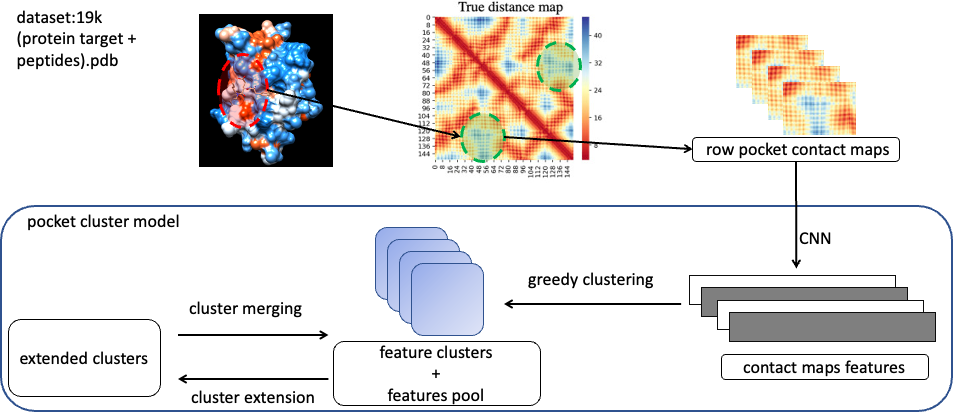
Figure1: Sampling of Pocking Domain
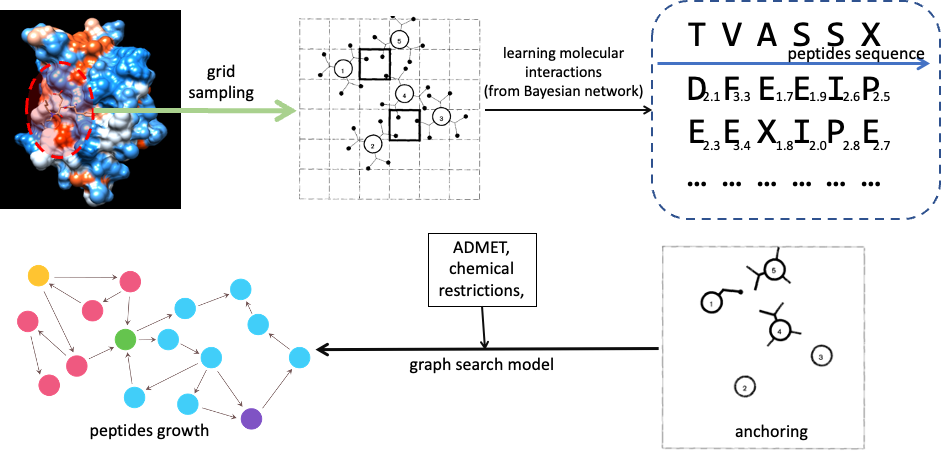
Figure2: Peptide generation
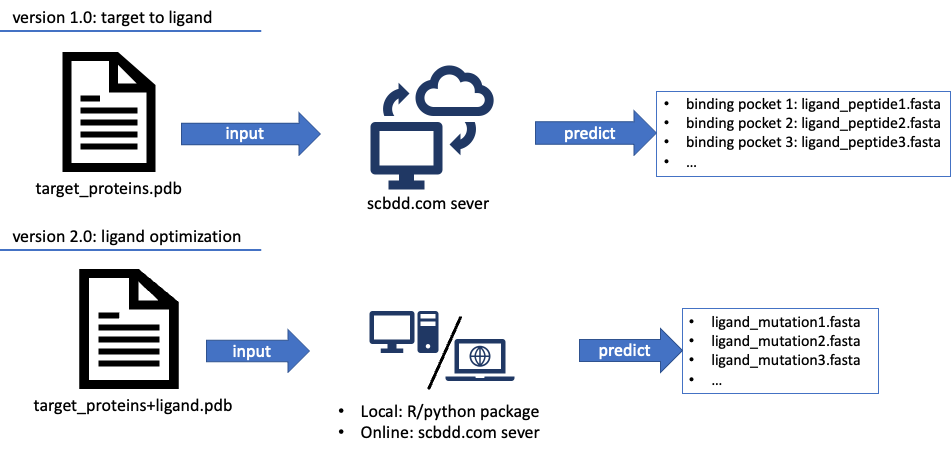
Figure3: We envisioned how-to-use
Week 4 : 2021/03/28
Figure4: Dr. Cao, our instructor, envisioned the base framework of Peplib Generator
Week 5 : 2021/04/05
We began with peptide-protein interaction prediction model, pre-processed PPI databases like Propedia and Pepbdb. We’ve also tested speed of peptide-protein docking, which is quite depressing for it took 9-13 hours.
Week 6 : 2021/04/11
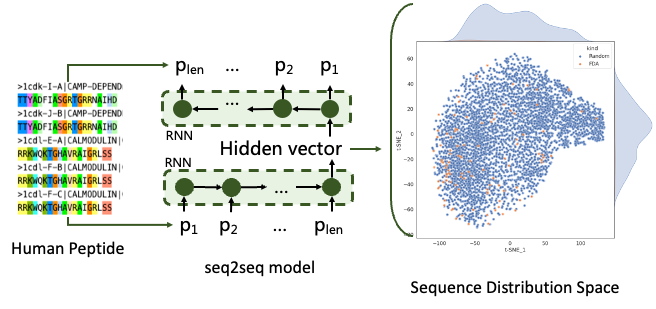
Figure5: Evolution Algorithm: a sequential genetic algorithm framework is proposed.
Week 7 : 2021/04/17
A transformer-based model is expected to be used on PPI model, we learnt how does multi-attention layer work.
Week 8 ~ Week 9
We suspended our project for Mid Term Exams.
Week 10 : 2021/05/09
We used an encoder-decoder system to represent peptide, a part of data from peptideatlas is used to train the model, reached a high performance in both 256 and 512 dimensions. We discussed how to represent PPI complexes to train PPI model, a structure-based dataset contains much more information in a data pair than a sequential-based one, yet to build a model to represent structure complex is a bigger challenge, and gladly we accepted it.
Figure6
Week 11 : 2021/05/16
A part of our team took apart in the 1st Biological Computing Conference of China. And adjusted project to avoid some overlap work for other Drug companies like XtalPi.
Week 12 : 2021/05/23
We built a framework from ResNet and StyleGAN as PPI prediction model.
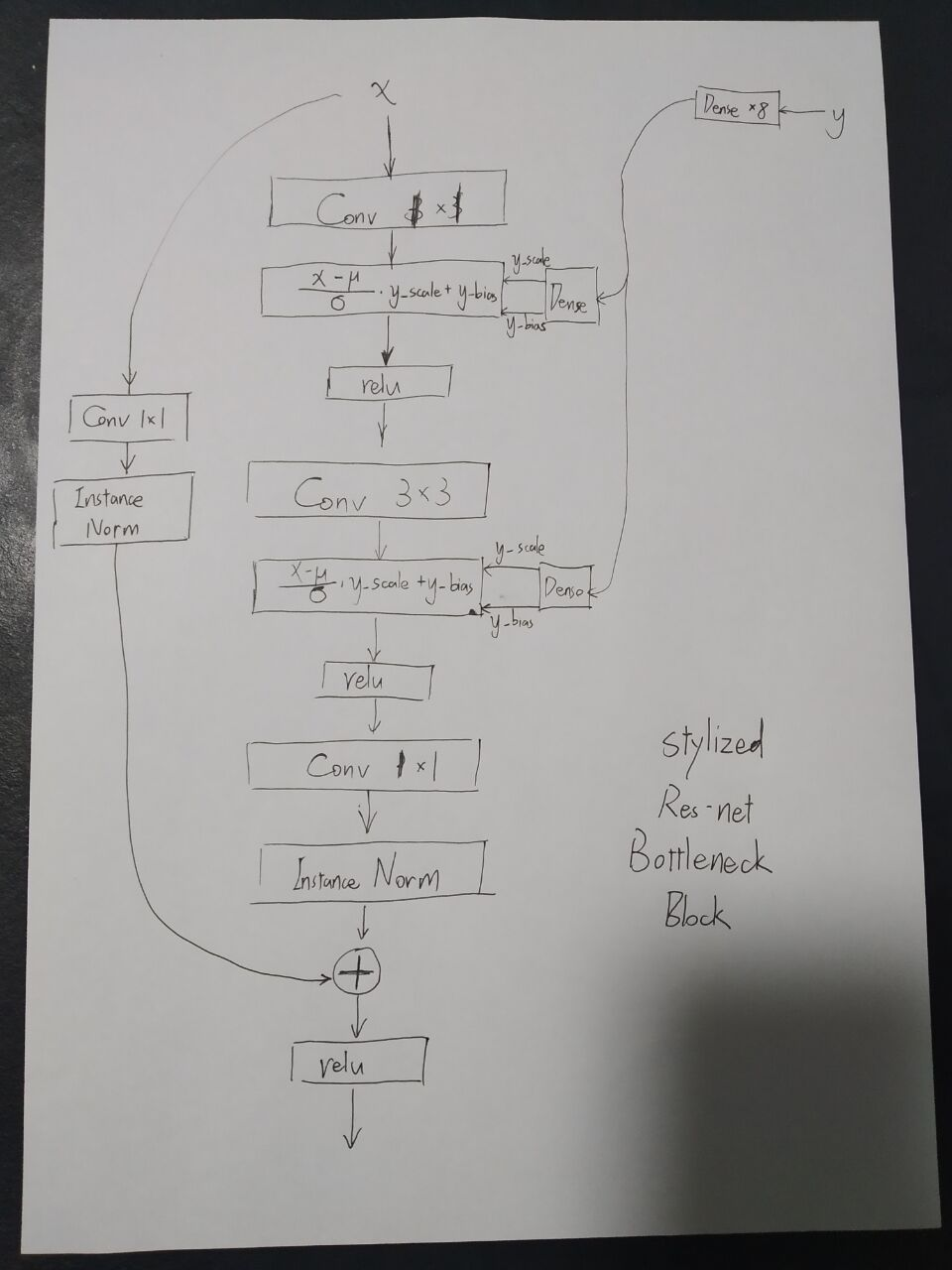
Figure7: PPI prediction model
Week 14 : 2021/05/31
We designed a UML diagram about how we deploy models in servers.
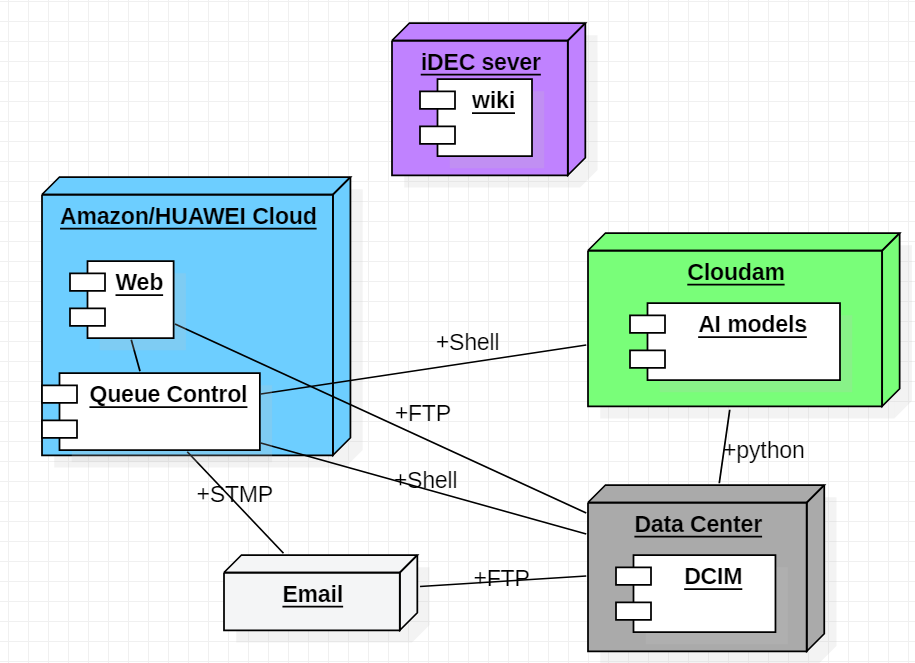
Figure8
Week 15 ~ Week 21
We suspended our project for Final Term Exams.
Week 22 : 2021/08/06
Successfully, our Evolution Algorithm worked. We tested its performance with peptide representation model, and it find the longest sequence(50aa) in only 18s. However, the 1st PPI prediction model based on ResNet failed as the accuracy of it float around 50%. Here’s a hypothesis to explain its low accuracy: model can learn nothing but noise from our poorly represented structural dataset. Based on the hypothesis, we find a Geometry-CNN powered representation method: masif.
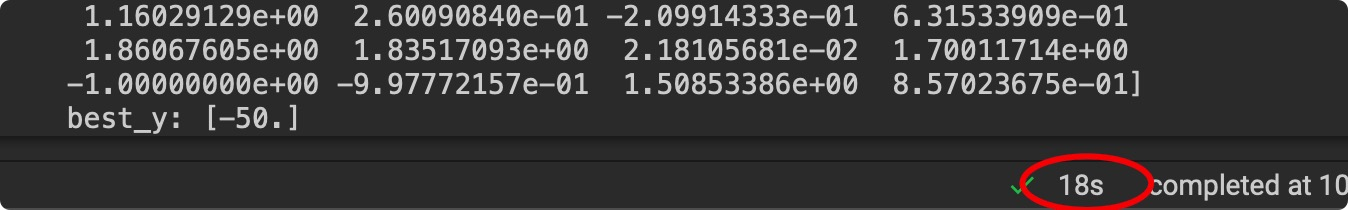
Figure9
Week 23 : 2021/08/15
We managed to deploy masif in the server, and represent all target protein using mesh data.
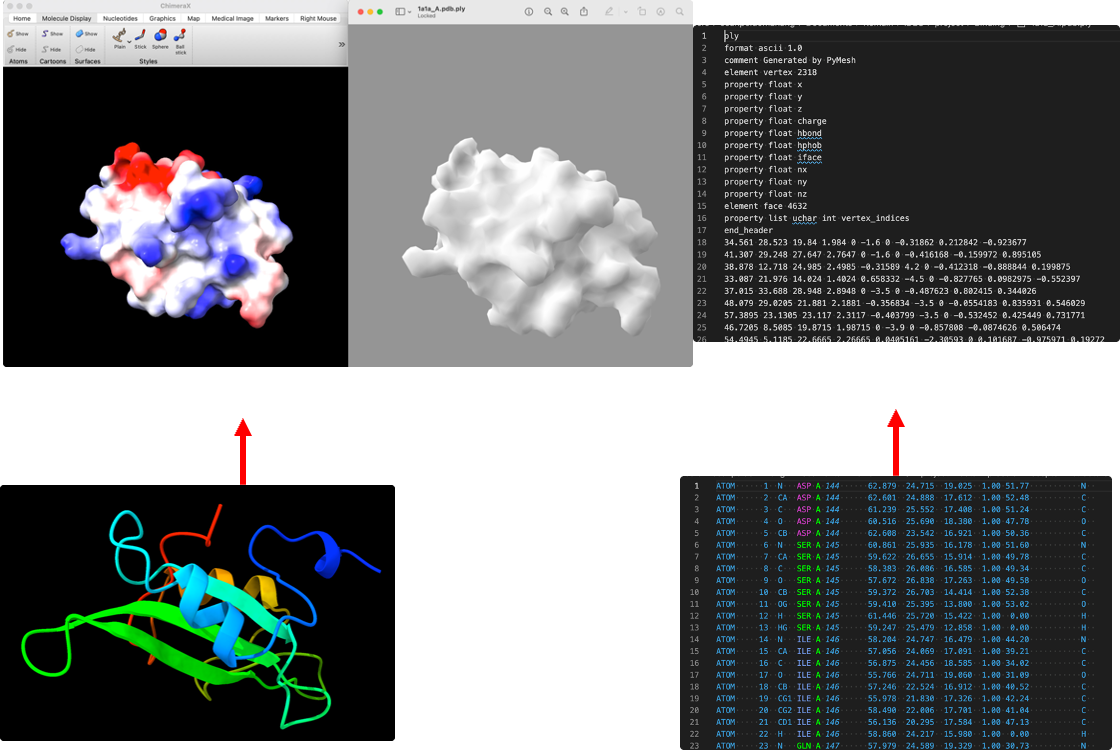
Figure10
Week 25 : 2021/08/23
We tried to embed mesh data into our PPI model.
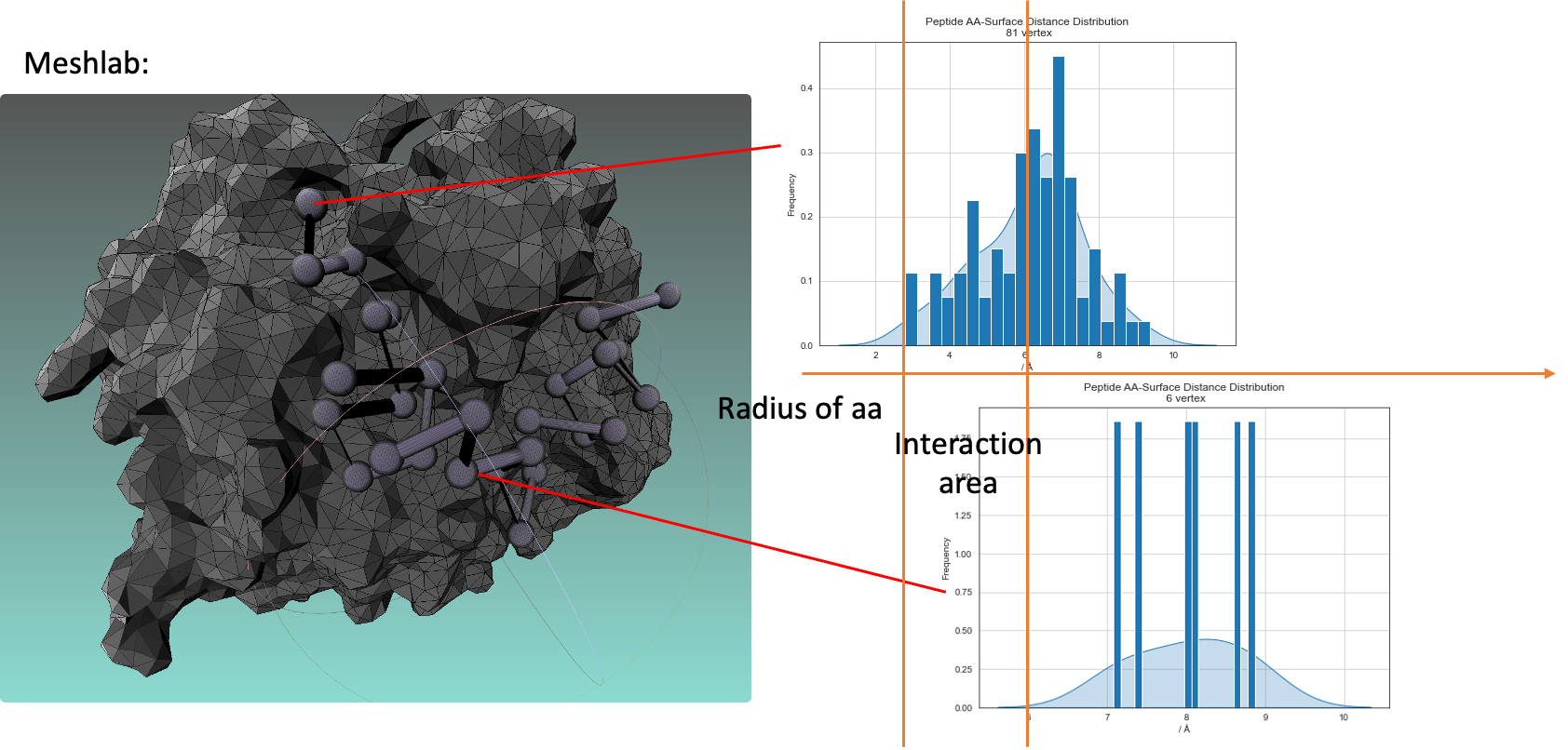
Figure11
Week 26 : 2021/08/30
We joined 8th-CCiC in Fudan University, Shanghai, China.
We tested performance of AlphaFold2 in peptide-protein interaction prediction.
Requirements of a good prediction result can be summarized as follows:
A peptide with consistent 2nd structure or other intrinsic interaction like disulfide bonds in complex conformation
A flat, stable and hydrophobic binding pocket

Figure12: A group photo of our team members
Week 27 : 2021/09/06
We updated Evolution Algorithm, added a group of new features.
Figure13
Week 28 : 2021/09/13
We accelerate AlphaFold2, renamed it to turbo-AF2. Combined with updated Evolution Algorithm, we tested Peplib Generator, optimized a peptide sequence for better stability:
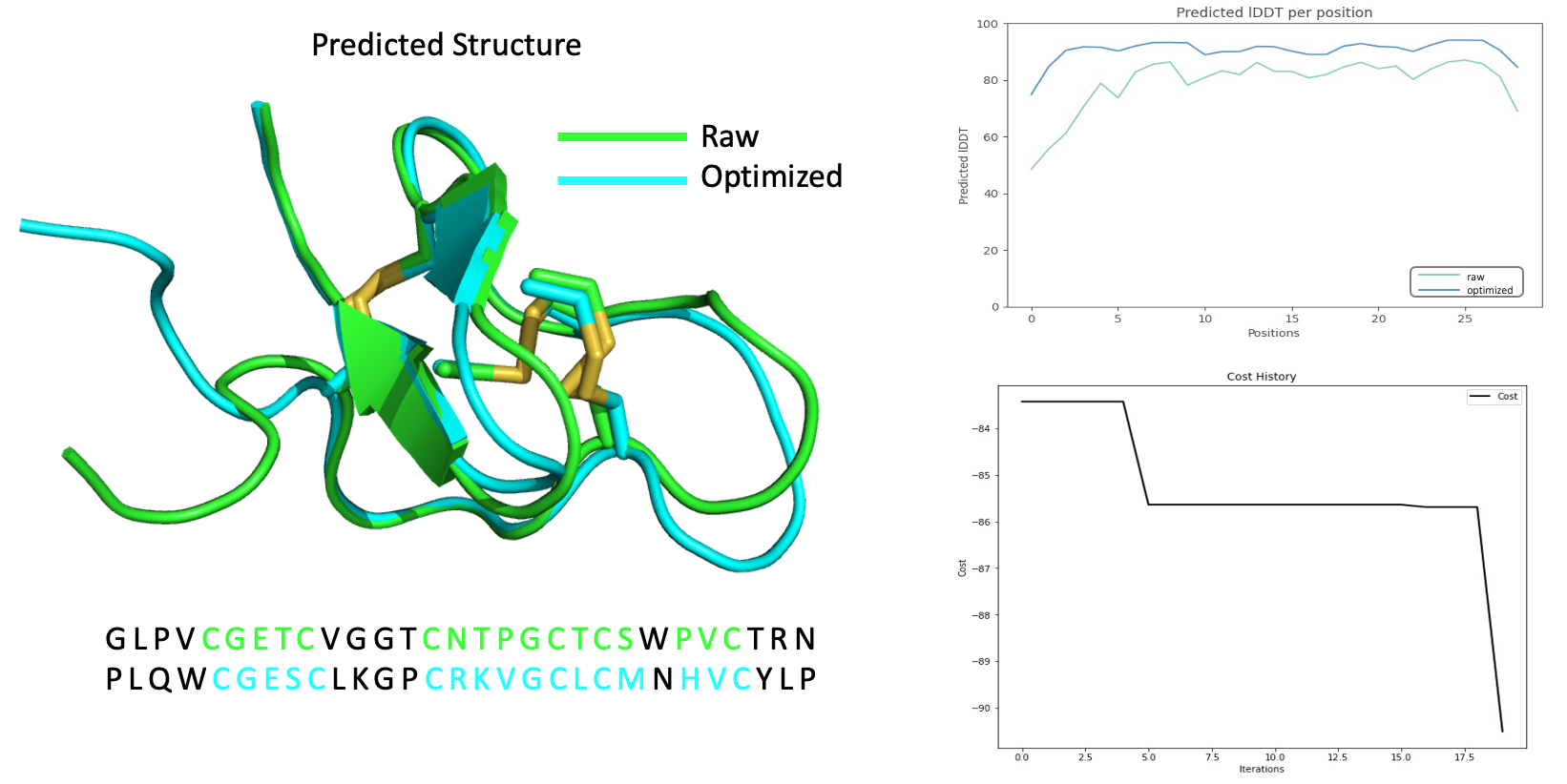
Figure14: Combined with updated Evolution Algorithm, we tested Peplib Generator, optimized a peptide sequence for better stability
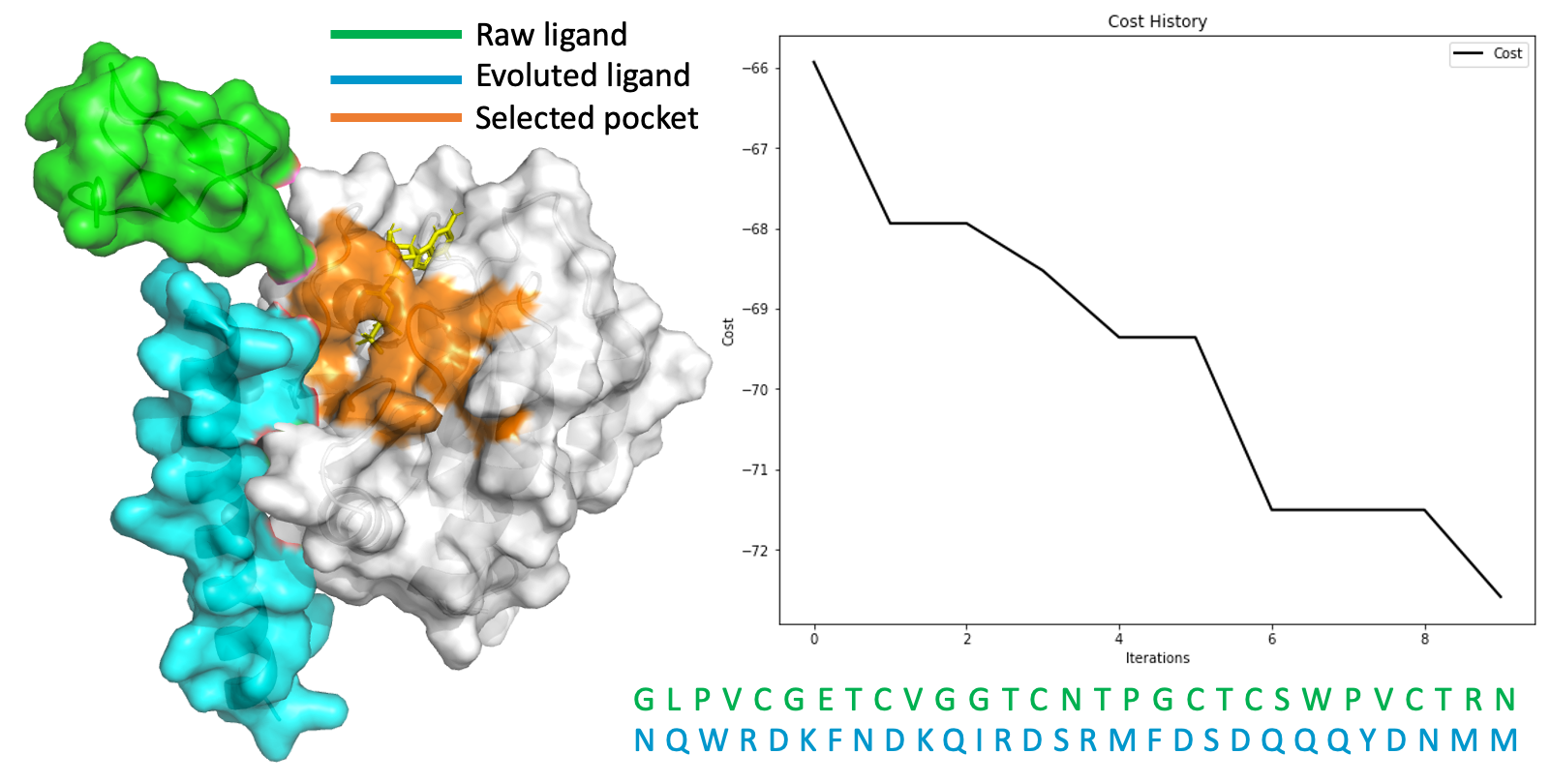
Figure15: Mutations seemed senseful, with grate excitement, we tested its ability in generating peptide ligand, but it failed
Week 29 : 2021/09/22
We update its performance in generate stable ligands, and used an easier target, successfully we nailed it.
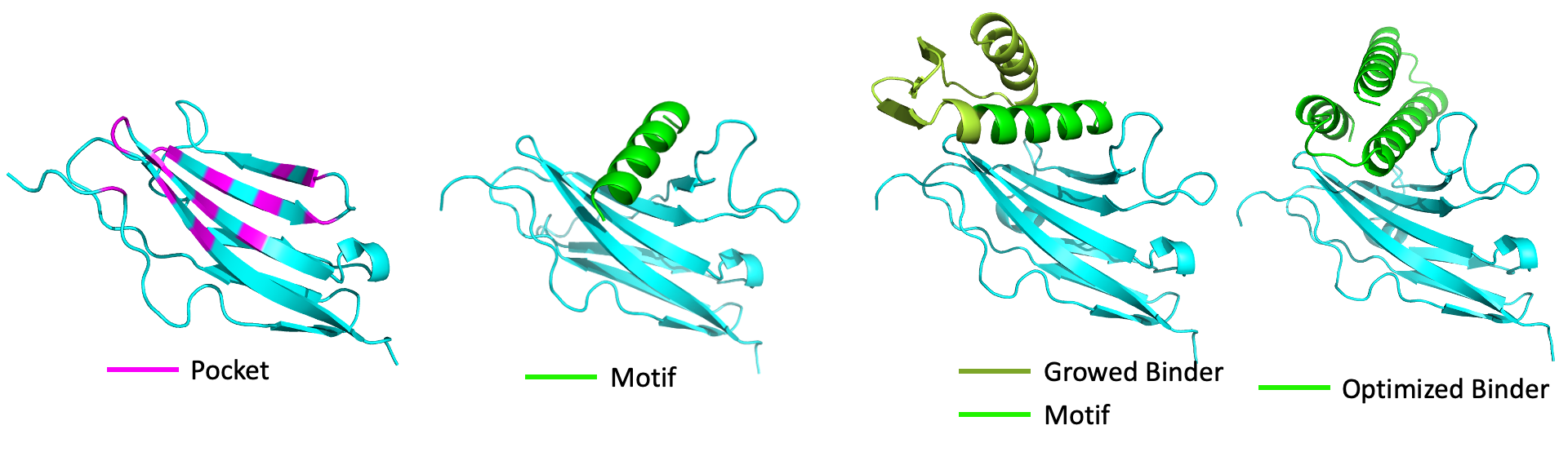
Figure16: A part of sequence data is provided in Supplementary
Week 30 : 2021/10/02
We kept updating its performance, and made more tests, got some conclusions about its pros and cons, they are concluded in our Reports.
Week 31 : 2021/10/10
We attended the 1st Synbiopunk in Shanghai, China. And make the length of generated peptide variable.

Figure17